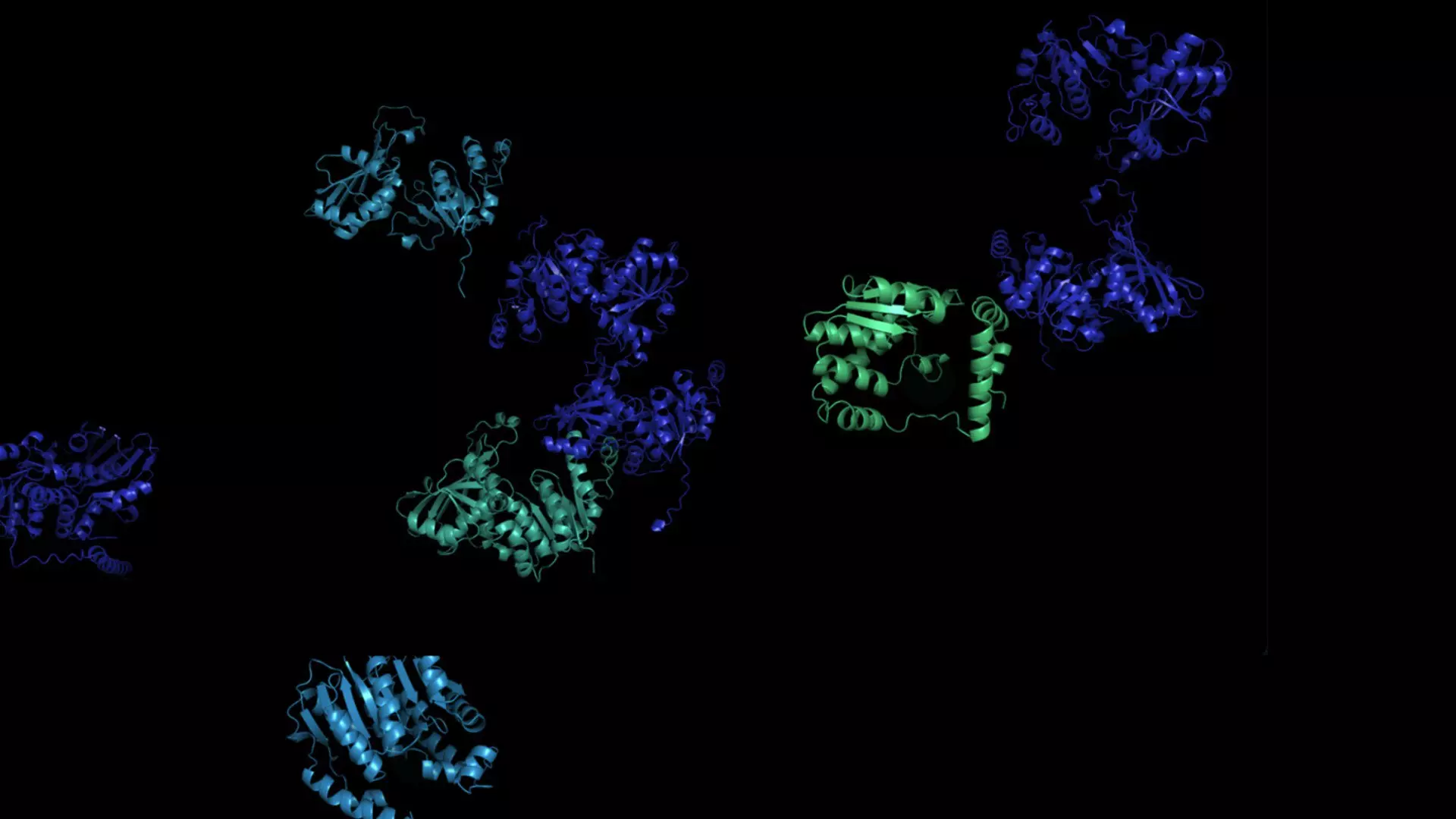
Amikor a londoni Deep Mind munkatársai idén nyáron mintegy 220 millió fehérje előre jelzett szerkezetét prezentálták, a DNS-adatbázisokban szereplő ismert szervezetek szinte minden fehérjéjét sikerült lefedniük. Most egy másik rendszer új adatbázissal állt elő, amely sötét anyagként tölti ki a fehérjék ismert univerzumát.
A Meta (korábban Facebook) kutatói egy mesterséges intelligencia segítségével körülbelül 600 millió, eddig nem jellemzett baktériumból, vírusból és más mikrobából származó fehérje szerkezetét jelezték előre. „Ezek olyan a struktúrák, amelyekről a nagyon keveset tudunk. Hihetetlenül titokzatos fehérjékről van szó, amelyek nagyszerű betekintést kínálnak a biológiába” – mondja Alexander Rives, a Meta fehérjékkel foglalkozó csapatának kutatási vezetője.
A kutatócsoport november 1-jén preprintben ismertette eredményeit, amelyeket egy úgynevezett nagy nyelvi modell segítségével kapott. Ez egy olyan mesterségesintelligencia-típus, amely alapesetben szövegfelismerést végez, vagyis néhány betűből vagy szóból képes megjósolni a szöveg hátralevő részét.
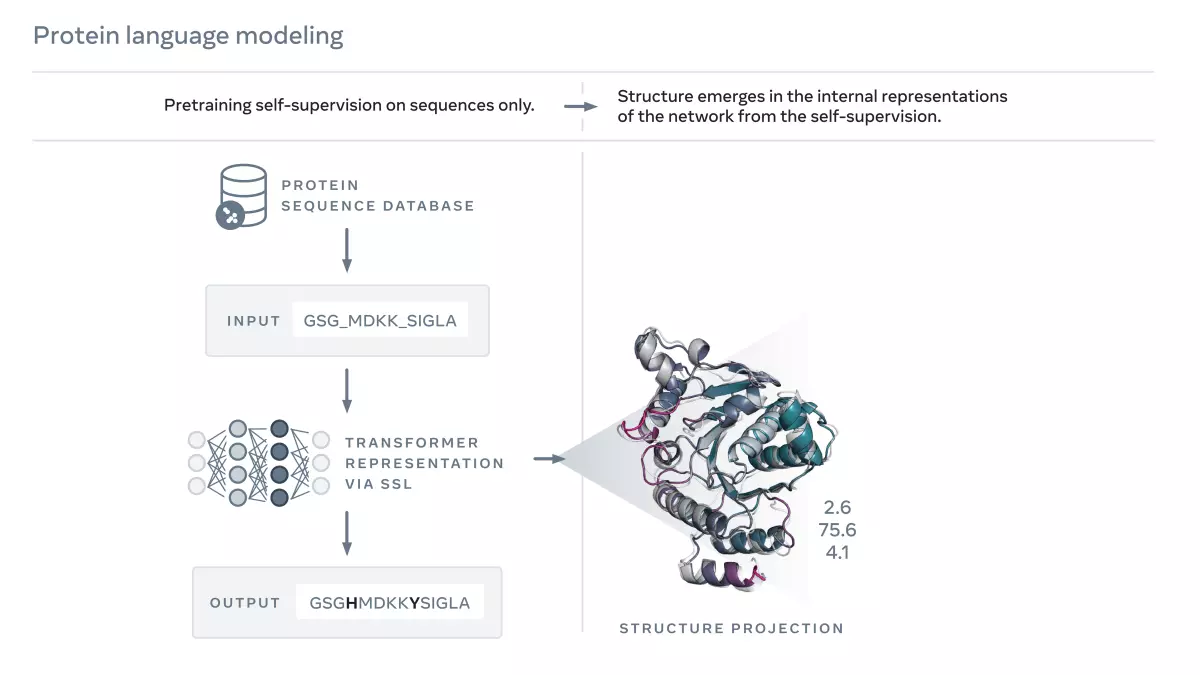
A hasonló nyelvi modelleket általában nagy mennyiségű szövegen képzik ki. Ahhoz, hogy a fehérjékre alkalmazhassák őket, Rives és kollégái ismert fehérjékhez tartozó szekvenciákat tápláltak a rendszerbe, amelyek 20 különböző aminosavból épülhetnek fel. A hálózat ezután megtanult automatikusan kiegészíteni olyan fehérjéket, amelyeknél az aminosavak egy részét kitakarták.
A tréning eredményeként rendszer képessé vált az aminosav-szekvenciák intuitív értelmezésére, ami azért érdekes, mert a szekvenciák a fehérjék térszerkezetével kapcsolatos információkat hordoznak, mondja Rives. A második lépésben ezeket az információkat hasznosították ismert fehérjeszerkezetek és szekvenciák közötti kapcsolatok kombinációja révén, így a rendszer képes volt a szekvenciákból struktúrákat előre jelezni.
A Meta ESMFold nevű rendszere nem olyan pontos, mint az AlphaFold, de Rives szerint saját rendszerük körülbelül 60-szor gyorsabb a szerkezetek előrejelzésében. Próbaképpen a modellt egy olyan adatbázisra alkalmazták, amely környezeti forrásokból – a talajból, a tengervízből, az emberi bélből, a bőrből és más mikrobiális élőhelyekről – származó, tömegesen szekvenált metagenomikus DNS-t tartalmaz. Az adatbázis elemeinek túlnyomó többsége olyan organizmusokból származik, amelyeket még soha nem tenyésztettek sikerrel laborban, és a tudomány számára gyakorlatilag ismeretlenek.
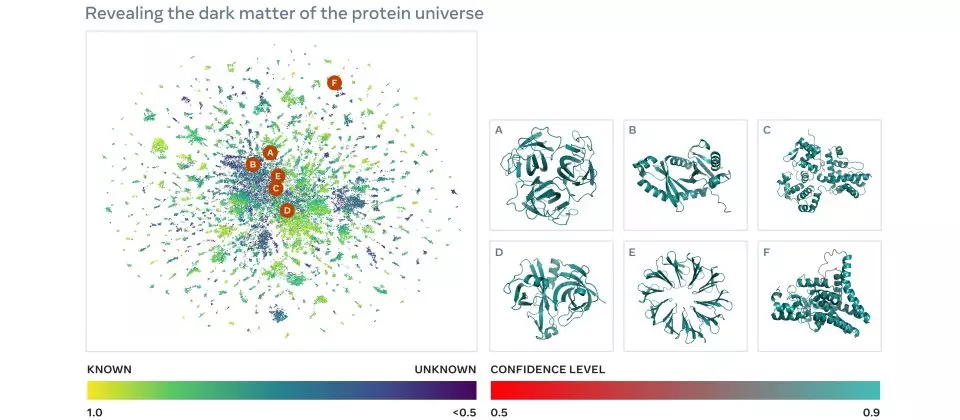
Az ESMFold összesen több mint 617 millió fehérje szerkezetét jósolta meg mindössze 2 hét alatt. Az előrejelzések és a modell alapjául szolgáló kód bárki számára szabadon hozzáférhető, mondja Rives. A 617 millió előrejelzés több mint egyharmadát ítélte jó minőségűnek a modell, vagyis ezek esetében a kutatók bízhatnak abban, hogy a fehérje általános alakja helyes, és egyes esetekben finomabb, atomi szintű részletek is pontosan felismerhetővé váltak.
A szerkezetek jelentős része teljes újdonság a kutatók számára, ezek nem hasonlítanak a kísérletileg meghatározott fehérjeszerkezetek adatbázisaiban vagy az ismert szervezetekből származó előrejelzéseket tartalmazó AlphaFold adatbázisában találhatóakhoz sem. Burkhard Rostot, a Müncheni Műszaki Egyetem kutatója arra is felhívja a figyelmet, hogy a nyelvi modellen alapuló előrejelzési módszerek kifejezetten előnyösek annak gyors meghatározására, hogy a mutációk hogyan változtatják meg a fehérjék szerkezetét, ami az AlphaFolddal nem lehetséges.
