1. oldal
A kémiai reakciók általában rendkívül gyorsan zajlanak le: elektronok helyeződnek át, kötések szűnnek meg és alakulnak ki, megváltozik a molekulák szerkezete, és mindez olyan sebességgel, meg persze olyan miniatűr méretekben történik, hogy élőben megfigyelni szinte lehetetlen, de legalábbis rendkívüli technikai erőfeszítést igényel. Napjainkban ugyan már lehetséges a molekulák átalakulását mikroszkóp alatt vizsgálni, ehhez azonban olyan speciális felszerelésre van szükség, amely a szakértők többségének nem áll rendelkezésére, arról nem is beszélve, hogy milyen költséges vállalkozás. Vannak azonban más utak is: 2013 kémiai Nobel-díjasai évtizedekkel ezelőtt kezdtek dolgozni azon, hogyan lehetne a kémiai reakciókat részleteikben nyomon követni számítógépes segítséggel.
Martin Karplus, Michael Levitt és Arieh Warshel munkásságának köszönhetően napjaink vegyészei jelentős részben számítógépen tervezik meg és futtatják le kísérleteiket. A három kutató a hetvenes években kezdett dolgozni azokon a módszereken, amelyek mára elengedhetetlenné váltak a kémia tudományának művelői számára. Hogy mindez megvalósítható legyen, a szakértőknek először is minden korábbinál aprólékosabban meg kellett vizsgálniuk a komplex kémiai folyamatok legkisebb lépéseit is, hogy aztán később szoftveresen modellezni tudják ezeket.

Michael Levitt, Martin Karplus és Arieh Warshel
Tegyük fel például, hogy tudósként a fotoszintézist szeretnénk tanulmányozni, mégpedig abból a célból, hogy minél tökéletesebben „lemásoljuk” a folyamatot, de legalábbis annak egyes részeit, jobb napelemek vagy éppen hatékonyabb üzemanyagcellák létrehozása reményében. Első lépésként a fotoszintézist vezérlő fehérjékkel akarunk közelebbről megismerkedni. Napjainkban ezek több online adatbázisban is szabadon hozzáférhetők, így számítógépünkre letöltve három dimenzióban vizsgálhatjuk a több tízezer atomból álló szerkezeteket. A fehérjék struktúrájában valahol ott rejtőzik a „lényeg”, vagyis a reakcióközpont, ahol a vízbontás zajlik. A tényleges reakcióban csak néhány atom vesz részt (többek közt négy mangán- és egy kalcium-ion, illetve néhány oxigén atom), azonban ezek térbeli elrendeződésén túl az általunk beszerzett szerkezeti ábrából nem sok fog kiderülni. Ebből nem fogjuk megtudni, hogy pontosan milyen szerepet töltenek be ezek az atomok, hogyan távoznak a víz molekulájáról az elektronok, és mi lesz a keletkező protonokkal.
A folyamat részleteit hagyományos kémiai módszerekkel lehetetlen lenne nyomon követni, hiszen a másodperc töredéke alatt mennek végbe, így kísérleti úton nem lehet ezeket megfigyelni, a proteinek szerkezete pedig önmagában nem jelent segítséget a megértésben, hiszen a képernyőn teljesen passzívak. A növények leveleiben azonban egyáltalán nem ilyen statikusak a dolgok: a napfény hatására a fehérjék energiát vesznek fel, atomi szerkezetük pedig megváltozik. Ahhoz pedig, hogy ezt az újfajta struktúrát megismerjük, már elő kell vennünk azon szoftverek egyikét, amelyek az idei év Nobel-díjasainak köszönhetik születésüket.
A program segítségével különféle lehetséges reakciós utak szimulálhatók, és ezeken keresztül a felhasználó képet alkothat arról, hogy pontosan mit is csinál az adott atom egy-egy részfolyamat során. A legvalószínűbbnek tűnő reakciósor azonosítása után pedig már jóval könnyebb olyan kísérleteket tervezni, amelyek révén igazolható, hogy valóban úgy zajlik-e a folyamat, ahogy azt a számítógép sejteni véli. A kísérletek eredményeinek felhasználásával aztán még pontosabb szimulációk állíthatók össze, így az elmélet és a gyakorlat kölcsönösen segíteni tudja egymást. A metódus elterjedésének köszönhetően napjaink kémikusai legalább annyi időt töltenek a számítógép előtt, mint kémcsövekkel bűvészkedve.

2. oldal
De mitől is olyan különleges Martin Karplus, Michael Levitt és Arieh Warshel programja? Különféle reakciókat szimuláló szoftverek már korábban is léteztek, ezek azonban kizárólagosan vagy a newtoni fizika, vagy a kvantummechanika elveire támaszkodtak. Mindkét változatnak akadnak előnyei és hátrányai is. A klasszikus fizikára alapozott szoftverek jól működnek, ha nagyméretű, bonyolult szerkezetű molekulákat akarunk nyugalmi helyzetben modellezni, pontosan kiszámítható, hogyan fognak elhelyezkedni az egyes atomok a térszerkezetben. Ez a fajta fizika azonban nem tud mit kezdeni az „élő” molekulákkal, vagyis azzal a helyzettel, amikor a reakcióba ténylegesen belépő molekula gerjesztett állapotba kerül.
A tényleges reakciók szimulálására ezért a kvantummechanikához fordultak a szakértők, vagyis ahhoz az elmélethez, amelyben az elektronok egyszerre képesek részecskeként és hullámként is létezni, és ahol Schrödinger híres, dobozba zárt macskája egyszerre élő és halott. A kvantummechanika elmélete jelenlegi ismereteink szerint a legjobb teória a körülöttünk lévő világ folyamatainak leírására, ennek megfelelően az ilyen szimulációk jóval realisztikusabbak is, mint a newtoni alapokon nyugvó szoftverek. Egyetlen probléma van ezekkel a programokkal: minden egyes feladat óriási számítási kapacitást igényel.
Egy fehérje minden atomjának és elektronjának kvantummechanikai szintű modellezése persze jóval részletesebb képet ad a molekuláról és annak lehetséges reakcióiról, ugyanakkor azonban olyan szintű számítástechnikai hátteret igényel, amilyenről pár évvel ezelőttig nem is álmodhattunk. A kvantummechanikai elveken nyugvó szimulációkkal a hetvenes években próbálkozó szakértők tehát csak nagyon apró molekulák reakcióit tudták ilyen módon vizsgálni, és a folyamatok modellezése közben teljesen figyelmen kívül kellett hagyniuk a reakciós környezetet, holott a való életben nagyon is lényeges, hogy milyen hőmérsékleten vagy éppen milyen oldatban találkoznak egymással a molekulák. Ha azonban ezeket a faktorokat is be akarták volna vonni a képbe, egy-egy reakció szoftveres lefuttatása évtizedeket vett volna igénybe.
A kétfajta megközelítés tehát meglehetősen eltérő eredményt hoz: a newtoni alapokon nyugvó szimulációk nem eléggé pontosak, a kvantummechanikai elvek szerinti modellek pedig kezelhetetlenül bonyolultak. Karplus, Levitt és Warshel azonban sikeresen megtalálta a középutat: munkásságuknak köszönhetően olyan modellek születtek, amelyekben sikeresen összekovácsolódnak a két szemléletmód előnyei.
A történet szálai a hetvenes évekig, Martin Karplus harvardi laboratóriumba nyúlnak vissza. Karplus a kémiai reakciók kvantummechanikai alapokon történő modellezésének híve volt, és munkatársaival ennek megfelelő működésű szoftverek fejlesztésén dolgozott. Arieh Warshel 1970-ben kezdett dolgozni a laborban, miután ledoktorált az izraeli Weizmann Intézetben. Doktoranduszként hozzáférése volt ez utóbbi intézmény GOLEM nevű számítógépéhez, amelyen Michael Levitt-tel közösen megírtak egy korszakalkotónak számító, klasszikus fizikai elveken alapuló szoftvert a molekulák modellezésére. A program révén a legnagyobb biológiai molekulák szerkezete is felvázolhatóvá vált.

3. oldal
Amikor Warshel a Harvardra érkezett, ezt a programot is magával vitte, majd kiindulási alapként használva Karplus segítségével rövidesen elkezdett dolgozni egy újfajta szoftveren, amely egészen másként közelítette meg a problémát, és a „megszokottól” eltérő elektronokra koncentrált. A molekulák többségében az elektronok egy-egy atommag körül vagy egy kötésben keringenek, akadnak azonban olyan molekulaszerkezetek is, amelyekben az elektronok egy része jóval szabadabban mozog több atommag közt. Ilyen, úgynevezett delokalizált elektronok találhatók például a retina működésében fontos szerepet játszó retinaldehidben is, amely molekula kvantummechanikai viselkedése régóta Karplus érdeklődésének középpontjában állt. Amikor ugyanis fény éri a retinát, a retinaldehid delokalizált elektronjai gerjesztett állapotba kerülnek, ennek hatására pedig megváltozik a molekula alakja, vagyis megtörténik a látás folyamatának első lépése.
Karplus és Warshel idővel sikeresen modellezte a retinaldehid működését, azonban ehhez először hasonló tulajdonságú, de jóval egyszerűbb szerkezetű molekulákkal kezdtek kísérletezni. Az általuk kidolgozott, 1972-re elkészülő szoftver kvantummechanikai elveket alkalmazott a delokalizált elektronok viselkedésének szimulálására, a többi elektron és az atommagok leírásához azonban a klasszikus fizika elveit használta fel. Egészen addig senki sem próbálkozott hasonlóval.
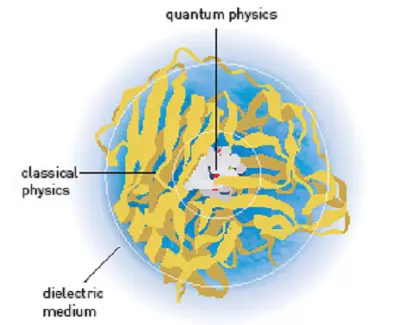
A mérföldkőnek számító programmal egyetlen probléma akadt: a molekulákat továbbra is csak nyugalmi állapotban lehetett vele szimulálni. Időközben azonban Michael Levitt és Warshel újra közös munkába kezdtek: elhatározták, hogy egy olyan szoftvert hoznak létre, amelynek révén az enzimek, vagyis az élő szervezetben lezajló kémiai reakciók legfőbb irányítóinak működése modellezhető. A cél elérése érdekében még szorosabb „együttműködést” kellett teremteni a klasszikus fizika és a kvantummechanika közt. Végül négy év fejlesztés után, 1976-ban mutatták be a kész programot, amely képes volt az enzimműködés szimulálására, és gyakorlatilag bármilyen kémiai reakció modellezésére − a molekulák méretétől függetlenül.
Napjaink kémiai szoftverei ugyanazokon az elveken alapulnak, amelyeket annak idején Karplus, Levitt és Warshel alkalmaztak: az elképzelés lényege, hogy oda kell koncentrálni a számítási kapacitást, ahol arra tényleg szükség van, vagyis a molekula azon elektronjainak és atommagjainak viselkedését érdemes kvantummechanikai alapokon modellezni, amelyek ténylegesen részt vesznek a reakció adott lépésében. A folyamatban érintetlenül maradó részeket pedig megfelelő pontossággal írják le a klasszikus fizika egyenletei is. A „spórolás” jegyében Levitt és Warshel még egy lépéssel tovább mentek: a számítógép nem követi folyamatosan figyelemmel a reakció szempontjából lényegtelen molekularészek minden egyes atomját, hanem ezeket tömbösítve, együtt kezeli.
A komplex kémiai reakciók számítógépes modellezése kulcsszerepet játszott és játszik ma is a különféle folyamatok megértésében. Karplus, Levitt és Warshel módszereinek legnagyobb erőssége abban rejlik, hogy univerzálisak: mindenféle kémiai folyamat tanulmányozható általuk a biológiai molekulák viselkedésétől kezdve a különféle vegyipari procedúrákig. És persze ugyanezek a metódusok bármikor tovább is fejleszthetők: Michael Levitt egy nemrégiben megjelent tanulmányában már egy teljes élő organizmus molekuláris szintű szimulációjának megvalósítási lehetőségeiről értekezik…